How to Run Molecular Dynamics Using SchNetPack
What is SchNetPack?
SchNetPack is a deep learning framework designed for atomistic simulations. You can use pre-trained models (like SchNet) to run molecular dynamics (MD) simulations that compute forces and energies on atoms using neural networks.
Requirements
- Python with SchNetPack installed
- ASE (Atomic Simulation Environment)
- A pre-trained model (e.g.,
md_ethanol.model) - A molecule file (e.g.,
md_ethanol.xyz)
Folder Structure
project-root/
├── md_configs/
│ ├── config.yaml
│ ├── dynamics/
│ ├── system.yaml
│ └── callbacks/
├── tests/testdata/
│ ├── md_ethanol.model
│ └── md_ethanol.xyz
├── src/scripts/spkmd.py
Configurations
In md_configs/dynamics/base.yaml, define:
n_steps: 1000000
defaults:
- integrator: md
In md_configs/integrator/md.yaml, define:
_target_: schnetpack.md.integrators.VelocityVerlet
time_step: 0.5
Run the Simulation
Use the following command (on Windows CMD) to start your simulation:
python spkmd.py \
simulation_dir=outputs/ethanol_test \
system.molecule_file=C:/full/path/to/md_ethanol.xyz \
calculator.model_file=C:/full/path/to/md_ethanol.model \
calculator.neighbor_list.cutoff=5.0
Output Explanation
- Runs for
n_steps= 1,000,000 - Each step = 0.5 fs → total time = 500,000 fs = 500 ps
- Trajectory saved every 10 steps = every 5 fs → 100,000 frames
- Output files in
outputs/ethanol_test/
Visualize Results
To view energy and temperature curves:
tensorboard --logdir outputs/ethanol_test/logs
Open http://localhost:6006 in your browser to explore the results.
Summary
This guide showed how to:
- Prepare your files and folders
- Configure Hydra and SchNetPack
- Run MD using a pre-trained neural network model
- Visualize the trajectory and energy logs
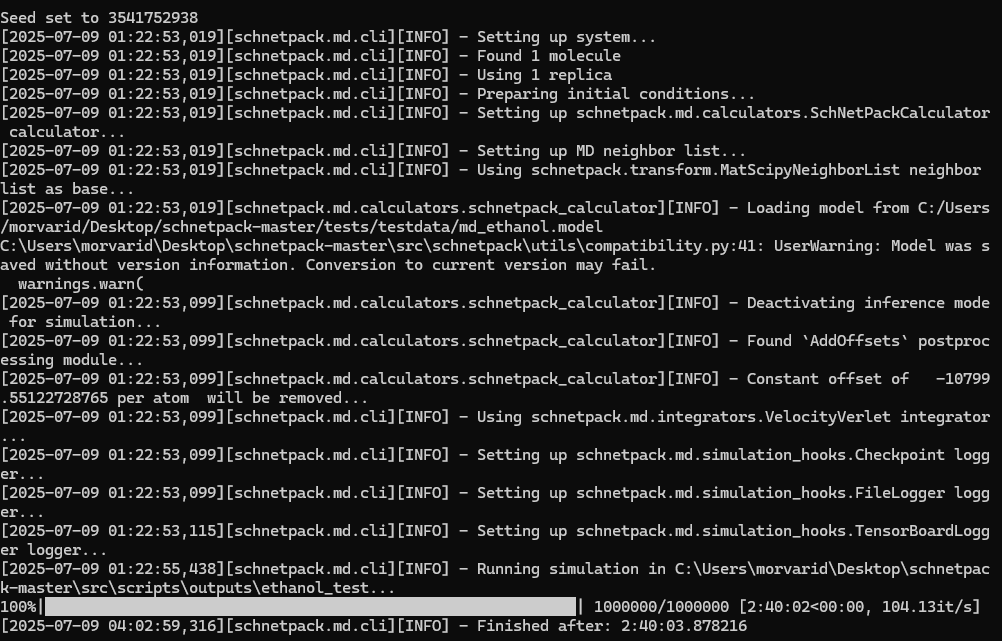
The image below shows a successful run of the SchNetPack MD simulation: